
09/09/2021
La citrulinemia tipo 1 es una de esas condiciones médicas que, aunque raras, subrayan la increíble complejidad del cuerpo humano y la importancia vital de la detección temprana. Se trata de un trastorno genético hereditario que afecta el ciclo de la urea, un proceso fundamental que nuestro organismo utiliza para eliminar el exceso de nitrógeno, un subproducto tóxico del metabolismo de las proteínas. Cuando este ciclo falla debido a la citrulinemia, el amoníaco se acumula en la sangre a niveles peligrosos, una condición conocida como hiperamonemia. Sin un diagnóstico y tratamiento inmediatos, esta acumulación puede causar daños neurológicos graves, coma e incluso la muerte en los primeros días o meses de vida. Este artículo profundiza en qué es la citrulinemia tipo 1, sus causas, síntomas, y lo más importante, las opciones de tratamiento que ofrecen una luz de esperanza a las familias afectadas.
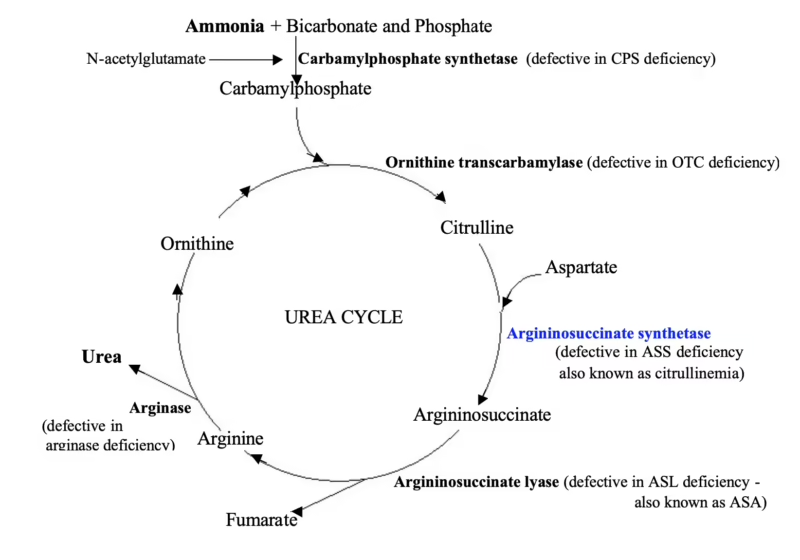
¿Qué es Exactamente la Citrulinemia Tipo 1?
Para entender la citrulinemia tipo 1, primero debemos hablar del ciclo de la urea. Piense en él como el sistema de purificación de su cuerpo. Cuando usted consume proteínas, su cuerpo las descompone en aminoácidos para usarlos en diversas funciones. El nitrógeno que no se utiliza debe ser eliminado de forma segura. El ciclo de la urea, que ocurre principalmente en el hígado, convierte este nitrógeno en urea, una sustancia mucho menos tóxica que luego se excreta a través de la orina.
Este ciclo consta de seis pasos enzimáticos. La citrulinemia tipo 1 es causada por una deficiencia en la tercera enzima de este proceso: la argininosuccinato sintetasa (ASS). Esta deficiencia se debe a mutaciones en el gen ASS1, que contiene las instrucciones para producir dicha enzima. Sin suficiente enzima ASS funcional, el ciclo se bloquea. La citrulina y, de manera más crítica, el amoníaco, se acumulan en el torrente sanguíneo, desencadenando una cascada de efectos tóxicos, especialmente en el cerebro, que es extremadamente sensible a los altos niveles de amoníaco.
Es una enfermedad autosómica recesiva, lo que significa que un niño debe heredar una copia del gen defectuoso ASS1 de cada uno de sus padres para desarrollar la enfermedad. Los padres, que solo tienen una copia del gen mutado, son portadores y generalmente no presentan síntomas.
Las Diferentes Caras de la Enfermedad
La citrulinemia tipo 1 no se presenta de una única manera. Su severidad y la edad de aparición de los síntomas pueden variar considerablemente, lo que lleva a clasificarla en varias formas:
- Forma clásica neonatal aguda: Es la forma más común y severa. Los bebés nacen aparentemente sanos, pero en los primeros días de vida comienzan a mostrar síntomas como letargo, rechazo a la alimentación y vómitos. La condición progresa rápidamente a edema cerebral, convulsiones y coma hiperamonémico si no se trata de inmediato.
- Forma de inicio tardío: En algunos individuos, los síntomas pueden no aparecer hasta la infancia tardía o incluso la edad adulta. Estos casos suelen ser más leves y los episodios de hiperamonemia pueden ser desencadenados por enfermedades, estrés o un alto consumo de proteínas.
- Forma especial en mujeres: Existe una variante que se manifiesta durante o después del embarazo, donde los cambios metabólicos y hormonales pueden desencadenar la enfermedad en mujeres que previamente eran asintomáticas.
- Forma asintomática: Algunas personas con la mutación genética pueden no desarrollar nunca síntomas, aunque esto es menos común.
Tabla Comparativa: Citrulinemia Tipo 1 vs. Tipo 2
Es importante no confundir la citrulinemia tipo 1 con la tipo 2, que es una enfermedad diferente causada por un defecto en un transportador de proteínas llamado citrina. A continuación, una tabla para diferenciar ambas condiciones.
| Característica | Citrulinemia Tipo 1 | Citrulinemia Tipo 2 |
|---|---|---|
| Causa Genética | Deficiencia de la enzima Argininosuccinato Sintetasa (gen ASS1). | Deficiencia del transportador de proteínas Citrina (gen SLC25A13). |
| Edad de Inicio Común | Neonatal (forma clásica) o en cualquier momento de la vida. | Generalmente en la edad adulta. |
| Síntomas Principales | Hiperamonemia, letargo, vómitos, coma, daño neurológico. | Hiperamonemia, síntomas neuropsiquiátricos (confusión, delirio), disfunción hepática. |
| Población Afectada | Distribución mundial, con mayor incidencia en poblaciones con alta consanguinidad. | Común en poblaciones de Asia Oriental. |
Síntomas y Señales de Alarma en Recién Nacidos
En la forma neonatal, los primeros días son cruciales. Un bebé con citrulinemia tipo 1 puede parecer perfectamente normal al nacer. Sin embargo, a medida que comienza a alimentarse (ya sea con leche materna o fórmula, ambas ricas en proteínas), el amoníaco empieza a acumularse. Las señales de alarma incluyen:
- Letargo progresivo: El bebé se vuelve cada vez más somnoliento y difícil de despertar.
- Rechazo a la alimentación: Pierde interés en comer.
- Vómitos: Puede vomitar después de las tomas.
- Irritabilidad: Llanto inconsolable.
- Problemas respiratorios: La respiración puede volverse rápida o, por el contrario, lenta e irregular.
- Hipotonía: Tono muscular deficiente, el bebé se siente "flojo".
- Convulsiones: A medida que aumenta la presión intracraneal.
Estos síntomas son una emergencia médica. La intervención inmediata es fundamental para prevenir daños cerebrales permanentes.
El Origen Genético: La Mutación p.Gly390Arg
Aunque se han identificado varias mutaciones en el gen ASS1, una de las más comunes y estudiadas es la mutación de sentido erróneo p.Gly390Arg (también descrita como c.1168G>A). Esta mutación específica ocurre en el exón 15 del gen y provoca la sustitución de un aminoácido (glicina) por otro (arginina) en una posición crítica de la enzima ASS. Esta alteración desestabiliza la estructura de la enzima, inactivándola casi por completo. La presencia de esta mutación en estado homocigoto (heredada de ambos padres) se asocia exclusivamente con la forma neonatal severa de la enfermedad. Estudios en diversas poblaciones, incluyendo Norteamérica, Europa y, como se ha visto en investigaciones recientes, en focos específicos de Oriente Medio, han identificado esta mutación como una de las principales responsables de la citrulinemia tipo 1, lo que la convierte en un objetivo clave para el diagnóstico genético.
Esperanza de Vida y Tratamientos Actuales
Aquí llegamos a la pregunta más importante: ¿cuál es la esperanza de vida? Sin tratamiento, el pronóstico es sombrío. Informes indican que la supervivencia más larga reportada para un bebé con la forma clásica no tratada fue de solo 17 días. Sin embargo, con un tratamiento rápido y continuo, el panorama cambia drásticamente.
El objetivo del tratamiento es doble: reducir los niveles de amoníaco en sangre lo más rápido posible durante una crisis y mantenerlos bajos a largo plazo para permitir un desarrollo lo más normal posible. Las estrategias incluyen:
- Restricción de Proteínas en la Dieta: La piedra angular del manejo a largo plazo es una dieta muy baja en proteínas para limitar la ingesta de nitrógeno. Esto debe ser cuidadosamente supervisado por un dietista metabólico para asegurar que el niño reciba las calorías y nutrientes esenciales para crecer.
- Suplementos de Aminoácidos: Se administra arginina o citrulina (dependiendo del defecto exacto del ciclo de la urea). En el caso de la citrulinemia tipo 1, la suplementación con arginina es vital. La arginina se convierte en un aminoácido esencial en estos pacientes y ayuda a estimular una vía alternativa para la excreción de nitrógeno.
- Fármacos Captadores de Nitrógeno: Medicamentos como el benzoato de sodio o el fenilbutirato de sodio se unen al amoníaco y otros precursores, creando compuestos que pueden ser eliminados del cuerpo a través de la orina, proporcionando una "vía de escape" para el nitrógeno.
- Manejo de Crisis Agudas: Durante un episodio de hiperamonemia severa, es necesario detener toda ingesta de proteínas y proporcionar altas dosis de calorías a través de glucosa intravenosa. En los casos más graves, se recurre a la hemodiálisis o hemodiafiltración, un procedimiento que filtra la sangre para eliminar rápidamente el amoníaco y otras toxinas.
Con este manejo integral, los niños pueden sobrevivir y llevar vidas plenas, aunque siempre requerirán un seguimiento médico y dietético estricto de por vida. El diagnóstico prenatal ofrece a las familias con antecedentes la posibilidad de conocer el estado del feto y prepararse para un tratamiento inmediato al nacer.
Preguntas Frecuentes (FAQ)
¿La citrulinemia tipo 1 tiene cura?
Actualmente no existe una cura definitiva para la citrulinemia tipo 1. Sin embargo, es una enfermedad manejable. El tratamiento es de por vida y se centra en el control dietético, la suplementación y la medicación para prevenir la acumulación de amoníaco y sus consecuencias tóxicas.
¿Si ambos padres son portadores, todos sus hijos tendrán la enfermedad?
No necesariamente. Cuando ambos padres son portadores de una enfermedad autosómica recesiva, en cada embarazo existe:
- Un 25% de probabilidad de que el hijo herede ambos genes mutados y tenga la enfermedad.
- Un 50% de probabilidad de que el hijo herede un gen mutado y sea un portador sano, como sus padres.
- Un 25% de probabilidad de que el hijo herede ambos genes normales y no tenga la enfermedad ni sea portador.
¿Cuál es el pronóstico a largo plazo con tratamiento?
El pronóstico ha mejorado enormemente con los avances en el diagnóstico y tratamiento. Los niños que son diagnosticados y tratados precozmente, antes de que ocurra un daño neurológico significativo, tienen muchas más posibilidades de tener un desarrollo intelectual y físico normal. Sin embargo, siguen estando en riesgo de sufrir crisis metabólicas, especialmente durante enfermedades o periodos de estrés, lo que requiere una vigilancia constante.
¿Por qué es tan importante la dieta?
La dieta es fundamental porque la fuente principal de nitrógeno en el cuerpo proviene de la descomposición de las proteínas que comemos. Al restringir estrictamente la ingesta de proteínas, se reduce la cantidad de "combustible" que puede convertirse en amoníaco tóxico, aliviando la presión sobre el ciclo de la urea defectuoso.
Conclusión: Un Futuro de Esperanza
La citrulinemia tipo 1 es un desafío formidable, una enfermedad que convierte un nutriente esencial como la proteína en una amenaza potencial. Sin embargo, es también un ejemplo del poder de la medicina moderna. Gracias a un profundo entendimiento de su base genética y metabólica, se han desarrollado estrategias que han transformado un pronóstico casi universalmente fatal en una condición crónica manejable. La clave reside en la sospecha temprana, el diagnóstico rápido y un equipo multidisciplinario dedicado que incluye genetistas, pediatras y dietistas. Para las familias que enfrentan este diagnóstico, el camino es exigente, pero está lleno de esperanza y la posibilidad de ver a sus hijos crecer y prosperar.
Si quieres conocer otros artículos parecidos a Citrulinemia Tipo 1: Esperanza de Vida y Claves puedes visitar la categoría Automovilismo.